

Collaboration and Data Sources
We received 120 samples of RNA extracted from birds that tested positive for West Nile virus from David Neitzel from the Minnesota Department of Health. 113 samples were selected and had suitable genome coverage after sequencing for phylogenetic analysis. Samples came from 39 counties and the years 2002-2005. In this update, we highlight the sequencing results and a few conclusions drawn from a maximum likelihood based phylogenetic analysis.
Data Generation
The sequencing data was generated using an amplicon-based sequencing scheme, PrimalSeq (Grubaguh et al. Genome Biology 2019). Our full protocol is available online here. Sequencing reads were aligned using bwa and processed using iVar.
Raw Data
Consensus sequences and BAM files along with associated metadata are available on Google Cloud. A summary of the alignment statistics, limited to samples collected in 2002, is shown below in Table 1. Statistics for the remaining samples are available on GitHub.
| Table 1. Alignment statistics | ||
|---|---|---|
| Sample | Percent Genome Coverage | Mean Coverage per Nucleotide |
| W1383 | 98.0% | 3265.7 |
| W1384 | 100.0% | 2619.4 |
| W1385 | 100.0% | 2548.7 |
| W1386 | 100.0% | 2201.0 |
| W1387 | 99.4% | 2416.4 |
| W1388 | 100.0% | 2798.9 |
| W1389 | 99.0% | 2640.7 |
| W1390 | 93.2% | 2425.0 |
| W1391 | 100.0% | 1953.4 |
| W1392 | 100.0% | 1389.0 |
| W1393 | 95.7% | 222.6 |
| W1394 | 100.0% | 1930.8 |
| W1395 | 100.0% | 2331.3 |
| W1396 | 99.9% | 430.0 |
| W1397 | 100.0% | 1824.1 |
| W1398 | 100.0% | 2490.6 |
| W1399 | 98.4% | 2065.4 |
| W1400 | 100.0% | 2588.2 |
| W1401 | 100.0% | 2586.2 |
| W1403 | 95.4% | 50.9 |
| W1404 | 92.4% | 1733.6 |
| W1407 | 96.9% | 1623.1 |
| W1408 | 100.0% | 2673.0 |
| W1409 | 100.0% | 2518.8 |
| W1411 | 99.5% | 3603.9 |
| W1412 | 100.0% | 2146.5 |
Preliminary Analysis
We constructed a maximum likelihood (ML) phylogeny using 1969 genomes of West Nile virus from USA including 113 of the provided samples which had genome coverage greater than 70%, highlighted in light blue. Tree and root to tip regression plot are shown in Figure 1.
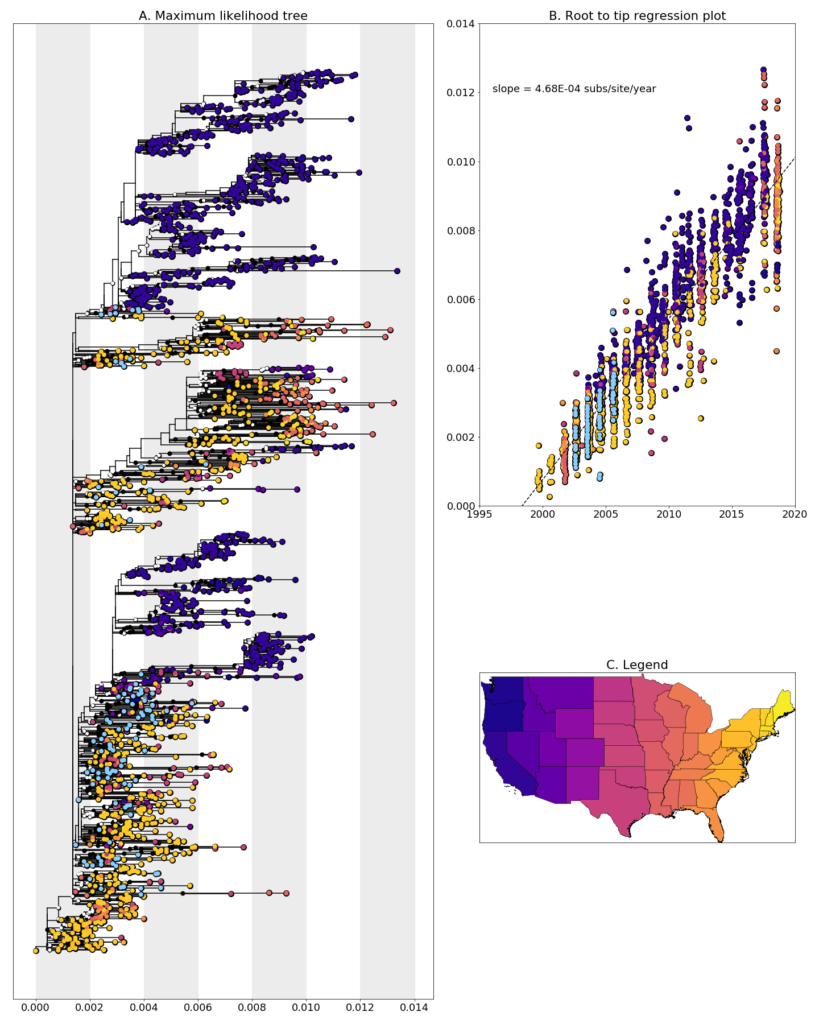
Figure 1: A. Maximum likelihood tree constructed using RAxML (bootstrap repeated 100 times) with 1969 genomes of West Nile virus from USA including 113 genomes provided by the Minnesota Department of Health (Highlighted in light blue). B. Root to tip regression plot. C. Legend
Minnesota sequences almost entirely fall within clades made up predominantly of sequences collected from the Eastern United States. The state which the sequences most closely lie with is New York which is unexpected given what we know about West Nile virus transmission, but likely a result of the undersampling of neighboring states. Besides New York, the most similar sequences are those from northern states such as North Dakota and Nebraska, however, a large amount of other states are also represented suggesting that the virus isn’t being geographically isolated. Long branch lengths and collection date differences make it difficult to conclusively determine the direction of virus movement.
Many transmission chains are evident in the state with sequences separating into numerous distinct clades. When sequences did cluster together they were often collected in the same year from disconnected counties, suggesting that the virus undergoes extensive mixing in the state. Although uncommon, we found nine clusters which provide evidence that West Nile virus transmission chains are able to persist from one year to the next in Minnesota. Interestingly, all of these clusters involved transmission into or out of Minneapolis and moved distinctly south to north. More sampling in the state, especially from 2006 onwards, is necessary to determine whether this is just a result of sampling bias or a real phenomenon.
Disclaimer
Please note that this data is released as work in progress by the WestNile 4K Project and should be considered preliminary. If you intend to include any of these data in publications, please let us know – otherwise please feel free to download and use without restrictions. We have shared this data with the hope that people will download and use it, as well as scrutinize it so we can improve our methods and analyses. Please contact us if you have any questions or comments.